Global alignment of protein interaction networks by graph matching methods
This site presents supplementary materials for the article "Global alignment of protein interaction networks by graph matching methods". It aims to propose a detailed description of the Results section presented in the paper.
Source data
In our experiments we used the data from http://www.cellcircuits.org/Bandyopadhyay2006/:
- Fly protein interaction network fly.intr with 7038 proteins (vertices), 20720 interactions (edges).
- Yeast protein interaction network yeast.intr with 4391 proteins (vertices), 14319 interactions (edges).
- Fly-Yeast InParanoid clusters table.SC-DM.highconfidence, 2244 clusters covering 3837 Fly and 2837 Yeast proteins.
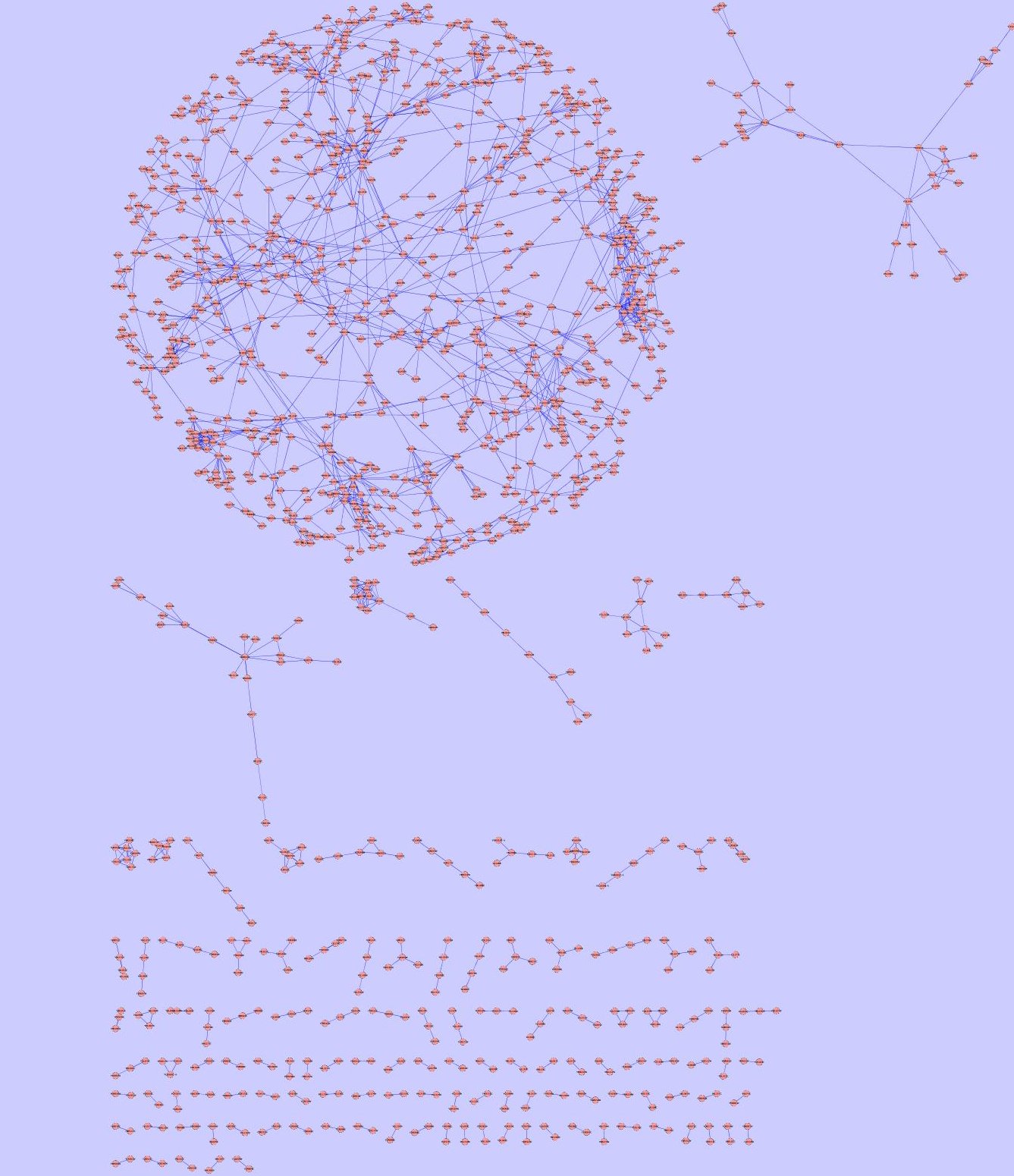
| Fly PPI network
|
Yeast PPI network
|

|

|
Up to date interaction data may be retrieved from the Database of Interacting Proteins server, InParanoid clustering software are available at http://inparanoid.cgb.ki.se/. We use the same date that was used by S.Bandyopadhyay and al. in order to be able to compare results of different algorithms.
Methods
- Markov random fields model (Systematic identification of functional orthologs based on protein network comparison, Sourav Bandyopadhyay, Roded Sharan and Trey Ideker)
- The IsoRank algorithm (Pairwise Global Alignment of Protein Interaction Networks by Matching Neighborhood Topology, Rohit Singh, Jinbo Xu and Bonnie Berger)
- The Gradient Ascent method (see paper, implemented in the GraphM package)
- The PATH algorithm (see paper, implemented in the GraphM package )
- The Message Passing algorithm (see paper, matlab script mp_algo.m)
Results.
Section 5.1 Disambiguation of functional orthologs within Inparanoid clusters
To check the number of conserved interactions produced by different matchings, you should download PPI network adjacency matrices fly_inp.adj and yeast_inp.ad, a matching of interest (with ids, the last column) and use the following script (octave, matlab, scilab)
f=load('fly_inp.adj');y=load('yeast_inp.adj');
G=zeros(max(f(:)));H=zeros(max(y(:)));
G(sub2ind(size(G),f(:,1),f(:,2)))=f(:,3);H(sub2ind(size(H),y(:,1),y(:,2)))=y(:,3);
T=load('mp_match.id');
num_cons_int=sum(sum(G(T(:,1),T(:,1)).*H(T(:,2),T(:,2))>0.25))/2
Section 5.2 Disambiguation of Inparanoid clusters with second-order interactions
To check the number of conserved interactions produced by different matchings, you can use the following script (octave, matlab, scilab)
f=load('fly_inp.adj');y=load('yeast_inp.adj');
G=zeros(max(f(:)));H=zeros(max(y(:)));
G(sub2ind(size(G),f(:,1),f(:,2)))=f(:,3);H(sub2ind(size(H),y(:,1),y(:,2)))=y(:,3);
T=load('path_match.id');
num_cons_int=sum(sum(G(T(:,1),T(:,1)).*H(T(:,2),T(:,2))>0))/2
Running algorithms
Here we show how results from previous section may be reproduced
Data preprocessing
Section 5.1
InParanoid clusters table.SC-DM.highconfidence provide information on protein pairs which may be assigned as functional orthologs. As it was mentioned already by S.Bandyopadhyay and al. in ``Systematic identification ...", the majority of Inparanoid clusters are trivial, consisting of two proteins (one of Fly, and one of Yeast), where the choice of functional orthologs is clear. Information on protein-protein interactions may be useful in ambiguous clusters with more than two proteins where some protein pairs may produce a conserved interaction with other pairs of proteins.
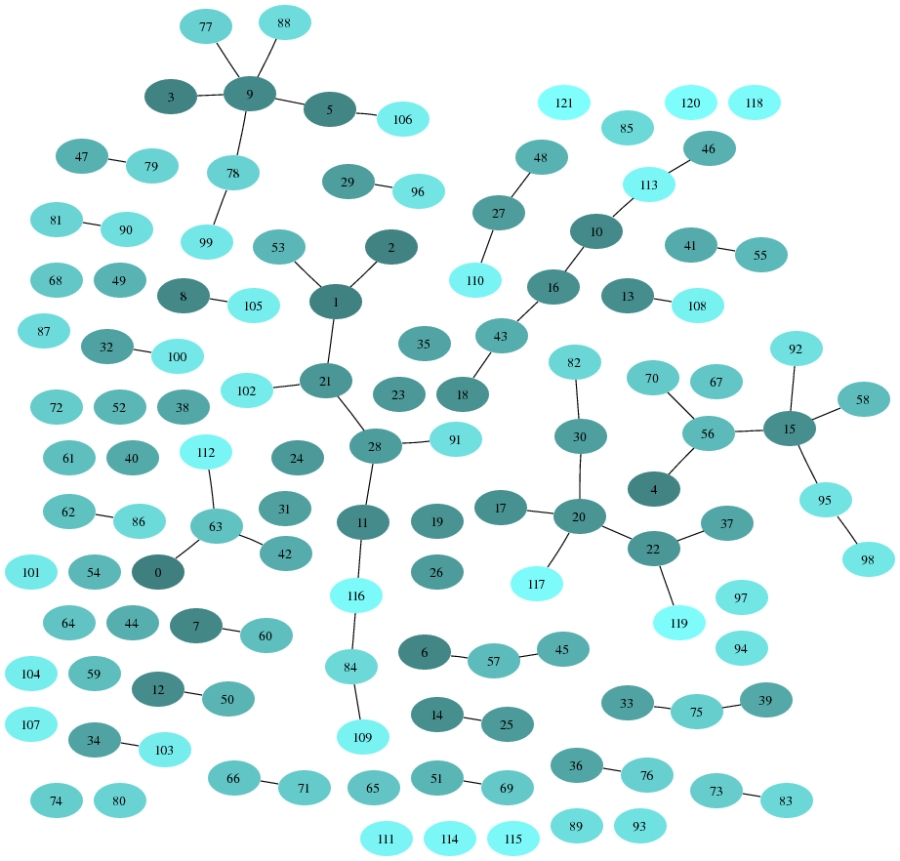
The graph on the figure below represents the system of InParanoid clusters
| InParanoid clusters
|

|
where each vertex corresponds to one cluster and there is an edge between two clusters C1 and C2 if there exists two pairs of proteins (f1,y1) in C1 and (f2,y2) in C2 such that one of the following conditions is true
- f1 interacts to f2, and y1 interacts to y2 in Fly and Yeast PPI networks correspondingly
- f1 interacts to f2, and y1 has a common neighbor with y2 in Fly and Yeast PPI networks correspondingly
- f1 has a common neighbor with f2, and y1 interacts to y2 in Fly and Yeast PPI networks correspondingly
Since we are interested only in clusters which are not trivial and which are not isolated (otherwise the information on PPI is useless), we keep only 121 InParanoid clusters.
| Ambiguous InParanoid clusters
|
|
Because we trace only ambiguous clusters, some clusters on the figure above are isolated, it means that they are connected to some trivial clusters and information on PPI may be useful for ortholog assignment in these clusters as well.
We can work directly with initial PPI networks, or we can consider only proteins which are interesting for ortholog assignment in ambiguous clusters of interest. After filtering out proteins which are not important, there stay 356 Fly proteins and 256 Yeast proteins.
At the end of the preprocessing step we obtain:
- ind_fly_sel (ids of fly selected proteins),
- ind_yeast_sel (idem for yeast),
- c_inp_sel.sim (matrix encoding Inparanoid clusters on selected proteins),
- fly_inp_sel.adj (ppi network of fly selected proteins),
- yeast_inp_sel.adj (idem for yeast).
Section 5.2
In this case we expand the definition of the conserved interaction by adding the forth condition.
- f1 has a common neighbor with f2, and y1 has a common neighbor with y2 in Fly and Yeast PPI networks correspondingly.
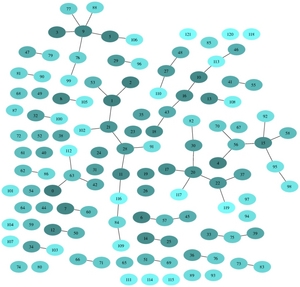
Because of the generalized definition, the graph of InParanoid clusters becomes much more complex (see figure below)
| Ambiguous InParanoid clusters (second-order interactions)
|

|
Now, there are 602 InParanoid clusters where information on PPI may be useful. After filtering out unimportant proteins there stay
- ind_fly_sel_so (ids of 984 fly selected proteins),
- ind_yeast_sel_so (ids of 736 yeast selected proteins),
- c_inp_so_sel.sim (matrix encoding Inparanoid clusters on selected proteins),
- fly_inp_so_sel.adj (ppi network of fly selected proteins),
- yeast_inp_so_sel.adj (idem for yeast).
Examples of scripts
To run the Message Passing Algorithm in matlab, you need to download (or create yourself) the following files: ind_fly_sel (Ids of Fly selected proteins), ind_yeast_sel (idem for Yeast), fly_inp_sel.adj (PPI network of Fly selected proteins), yeast_inp_sel.adj (idem for Yeast),c_inp_sel.sim (matrix encoding InParanoid clusters on selected proteins), trivial protein matchings ind_simple_match, mp_algo.m(matlab script implementing Message Passing). Then you just run the following code in matlab
%Message passing
T=mp_algo('fly_inp_sel.adj','yeast_inp_sel.adj','c_inp_sel.sim','ind_fly_sel','ind_yeast_sel','ind_simple_match');
%number of conserved interactions
f=load('fly_inp.adj');y=load('yeast_inp.adj');
G=zeros(max(f(:)));H=zeros(max(y(:)));
G(sub2ind(size(G),f(:,1),f(:,2)))=f(:,3);H(sub2ind(size(H),y(:,1),y(:,2)))=y(:,3);
T=load('path_match.id');
num_cons_int=sum(sum(G(T(:,1),T(:,1)).*H(T(:,2),T(:,2))>0.25))/2
The GA,PATH and IsoRank algorithms are implemented in the GraphM package. Here you have to provide PPI network adjacency matrices and matrix C encoding InParanoid clusters. Consider, for example, the case of second-order interactions (see Section 5.2 in the article). First, again, you download (or create yourself) ind_fly_sel_so (ids of 984 fly selected proteins), ind_yeast_sel_so (ids of 736 yeast selected proteins),c_inp_so_sel.sim (matrix encoding Inparanoid clusters on selected proteins),fly_inp_so_sel.adj (ppi network of fly selected proteins), yeast_inp_so_sel.adj (idem for yeast), trivial protein matchings ind_simple_match, and configuration file config.txt.
To run, for instance, the GA algorithm
./graphm config.txt "graph_1=fly_inp_so_sel.adj s;graph_2=yeast_inp_so_sel.adj s;C_matrix=c_inp_so_sel.sim s;algo=GA s;exp_out_file=ga_out.txt s;"
Here GA may be substituted by PATH, IsoRANK or any other graph matching algorithm from the GraphM package, for more information on algorithm parameters, see documentation of GraphM.
To count the number of conserved interactions, you can use graphm_nci.m (matlab script)
n=graphm_nci('ga_out.txt','fly_inp.adj','yeast_inp.adj','ind_fly_sel_so','ind_yeast_sel_so','ind_simple_match');